医療コラム
About Regulatory Approval of Medical Devices in Germany and the EU
daniel
Even if I am no lawyer I have some insights on the regulatory approval of medical devices in the EU since I am a German biomedical engineer. I want to share my knowledge by giving here a very brief introduction of the regulatory affairs involved with the development of medical devices in Germany.
Motivation:
We all know medical devices are used to treat people who have a disease, to heal and not to harm a patient. But many medical devices need to do harm in order to do “good”, e.g. a scalpel used to cut out a tumour or a CT scanner which sends radiation through your body to create a picture.
To measure how much harm is tolerable (risk) to cure a disease (benefit) is not up to the manufacturer, health care professional or patient. Because the manufacturer is greedy, the health care professional just wants to use the newest medical devices to publish in a medical journal and the patient is either being easily influenced or even has no other choice because of the lack of money.
Therefore society, or the governmental bodies and authorities elected by society, have to be involved to secure that every patient has the same right to receive treatment by safe and effective medical devices. This is guaranteed by a scheme of laws, decrees, directives and other forms of standards (statutory requirements).
The directives:
Europe consists of many different countries which each has their own idea of standards and norms. These standards are of course designed for the national approval scheme of the own country. So it is very expensive and complicated to design a medical device which approves all the different standards which are all in different languages and so on. This hinders innovation and leads to national isolation.
Therefore the European Commission revoked any use of technical standards and implemented one unified European approval scheme.
The so called directives were created. A directive in this sense is a legislative act of the European Union. The way of how the member states go conform with this directive is not dictated by the EU, only the result, e.g. safe and effective devices, is defined.
Each directive addresses a specific device category. As for example the MDD is the Medical Devices Directive and addresses therefore medical devices only. There is also a special directive for implantable medical devices.
Each directive has an Annex which provides a set of essential requirements for the safe and effective performance of the devices.
The manufacturer must use a so called “conformity assessment procedure” (CAP) to verify and demonstrate that his medical device complies with theses essential requirements.
The European Commission defined a number of modules which can be used for the various phases of the conformity assessment procedure.
The conformity assessment procedure must include a post production surveillance scheme for the notification of any adverse events of the device to the authorities.
Depending on the harm the medical device may produce different modules have to be used. It is obvious that a wood stick used by a dentist to hold the tongue down does not use the same conformity assessment procedures as for example a complex medical device like a CT scanner with its high voltage power source and X-ray radiation.
The modules can also be found in the Annex of the device specific directive.
Third party inspection/surveillance:
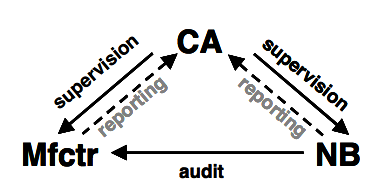
Source: Lecture Material on Regulatory Affairs by Prof. Dr. E. Schwanbom University of Applied Sciences Lübeck
Depending on the risks (to harm) involved with the use of the medical device, the manufacturer must allow a third party inspection/surveillance of his product and/or organisation by a so called “notified body”.
The notified body is typically an independent Non Governmental Organization (e.g. TÜV Rheinland AG) with the necessary qualification to audit (check) the compliance with the prescribed conformity assessment procedures and therefore with the essential requirements of the directive.
A competent authority (CA) is supervising the manufacturer (Mfctr) and the notified body (NB).
Non-compliance with the directives are being reported to the competent authority or to the EU Commission.
The competent authority is a governmental authority e.g. the BfArM in Germany.
If all essential requirements are met and the conformity assessment procedures are correctly implemented and checked by the notified body if necessary, the CE mark may be applied and the product is ready to be put on the market in the EU.
Summary:
The EU Commission revoked all technical standards on medical devices and created one directive for all member states of the EU to support innovation and make it easier to sell products in the EU.
Depending on the risk involved in the use of the medical device, specific conformity assessment procedures are used to go conform with the essential requirements and an inspection by a notified body may be necessary. If everything complies with the essential requirements and the conformity assessment procedures are correctly implemented a CE mark may be applied and the medical device can be freely marketed in the EU.
- Tweet
-

 2023/12/01
2023/12/01 2019/12/10
2019/12/10 2017/06/21
2017/06/21 2017/06/02
2017/06/02 2013/05/14
2013/05/14 2012/05/22
2012/05/22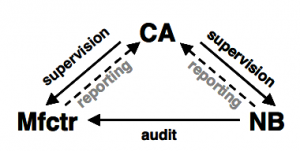 2012/03/07
2012/03/07 2011/11/14
2011/11/14